Section: New Results
Features Extraction
Axon extraction from fluorescent confocal microscopy images
Participants : Alejandro Mottini, Xavier Descombes, Florence Besse.
It it known that the analysis of axonal topologies allows biologists to study the causes of neurological diseases such as Fragile X Syndrome and Spinal Muscular Atrophy. In order to perform the morphological analysis of axons, it is first necessary to segment them. Therefore, the automatic extraction of axons is a key problem in the field of neuron axon analysis.
For this purpose, biologists label single neurons within intact adult Drosophila fly brains and acquire 3D fluorescent confocal microscopy images of their axonal trees. These images need to be segmented.
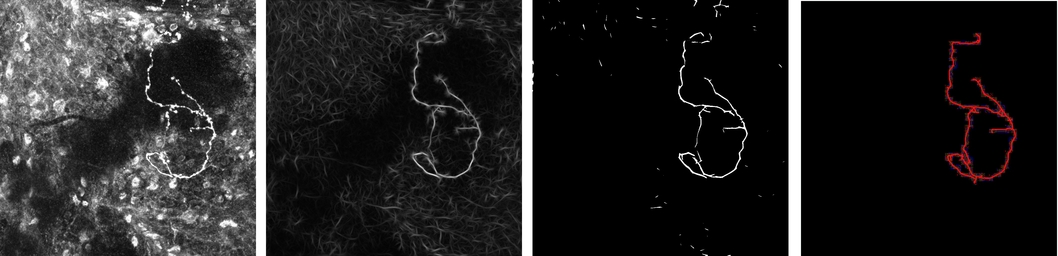
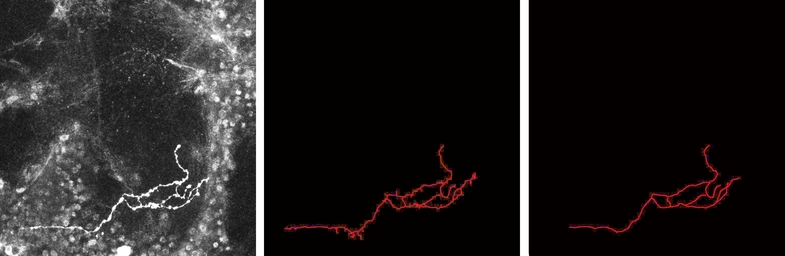
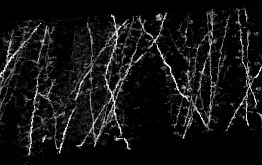
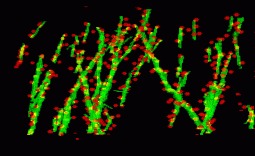
In our work presented in [16] , we propose a new approach for the automatic extraction of axons from fluorescent confocal microscopy images which combines algorithms for filament enhancement, binarization, skeletonization and gap filling in a pipeline capable of extracting the axons containing a single labeled neuron. Unlike other segmentation methods found in the literature, the proposed is fully automatic and designed to work on 3D image stacks. This allows us to analyze large image databases.
The method performance was tested on 12 real 3D images and the results quantitatively evaluated by calculating the RMSE between the tracing done by an experienced biologist and the automatic tracing obtained by our method. The good results obtained in the validation show the potential use of this technique in helping biologists for extracting axonal trees from confocal microscope images (see figures 5 and 6 ).
|
|
Dendrite spine detection from X-ray tomographic volumes
Participants : Anny Hank, Xavier Descombes, Grégoire Malandain.
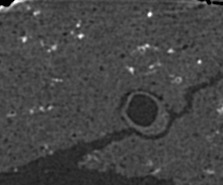
We have developped an automated algorithm for detecting dendritic spines from XRMT data. XRMT data allows imaging a large volume of tissue, and therefore a higher number of spines than laser scanning microscopy. We have shown that despite the lower image quality compared to microscopic data, we were able to extract dendritic spines. The main idea of the proposed approach is to define a mask for performing the spine detection without facing the false alarms problem as we introduce some information on spines localization. We therefore first extract the dendrites themselves and then compute the spine mask based on prior knowledge on their distance to dendrites. To extract dendrite we first compute the medial axis thanks to a multi-scale Hessian-based method. Then, we extract segments by a 3D Hough transform and reconstruct the dendrites using a conditional dilation. The spine mask is defined nerby the detected dendrites using anatomical parameters described in the literature. A point process defined on this mask provides the spine detection.
To exemplify the proposed approach, a subvolume (220 × 180 × 100) has been extracted from a XRMT volume that is given on figure 7 . As expected, the spines appear as small objects, whose size is close to the image resolution, along the tubular structures representing dendrites. Using the localization information to detect spine is essential to prevent false alarms due to noise or to the deviation of dendrites from a cylinder model. Figure 7 shows the detected dendrite medial axis and the obtained spine detection. The obtained results are promising and correspond to a visual inspection of the data. Forthcoming validation study will allow to better assess the quality of the detection by providing a quantitative evaluation.
Cell detection
Participant : Xavier Descombes.
This work was done in collaboration with Emmanuel Soubies and Pierre Weiss from ITAV (Toulouse)
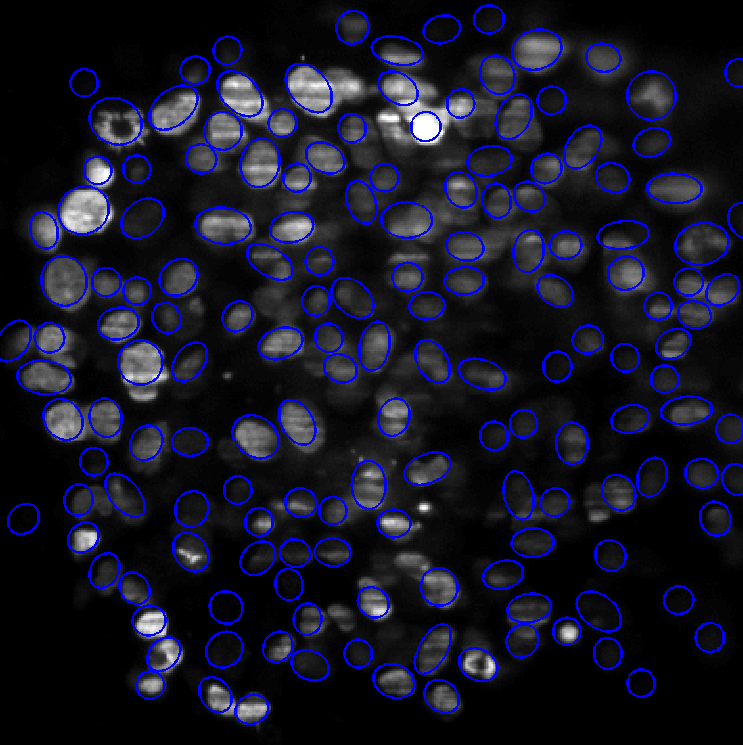
We have proposed some improvements of the Multiple Birth and Cut algorithm (MBC) in order to extract nuclei in 2D and 3D images. We have introduced a new contrast invariant energy that is robust to degradations encountered in fluorescence microscopy (e.g. local radiometry attenuations). Another contribution of this work is a fast algorithm to determine whether two ellipses (2D) or ellipsoids (3D) intersect. Finally, we propose a new heuristic that strongly improves the convergence rates. The algorithm alternates between two birth steps. The first one consists in generating objects uniformly at random and the second one consists in perturbing the current configuration locally. Performance of this modified birth step is evaluated and examples on various image types show the wide applicability of the method in the field of bio-imaging.
Figure 8 left shows the segmentation result on a Drosophila embryo obtained using SPIM imaging. This is a rather easy case, since nuclei shapes vary little. The images are impaired by various defects: blur, stripes and attenuation. Despite this relatively poor image quality, the segmentation results are almost perfect. The computing time is 5 minutes using a C++ implementation. The image size is 700 × 350. Figure 8 right presents a more difficult case, where the image is highly deteriorated. Nuclei cannot be identified in the image center. Moreover, nuclei variability is important meaning that the state space size χ is large. Some nuclei are in mitosis (see e.g. top-left). In spite of these difficulties, the MBC algorithm provides acceptable results. They would allow to make statistics on the cell location and orientation, which is a major problem in biology. The computing times for this example is 30 minutes.
|

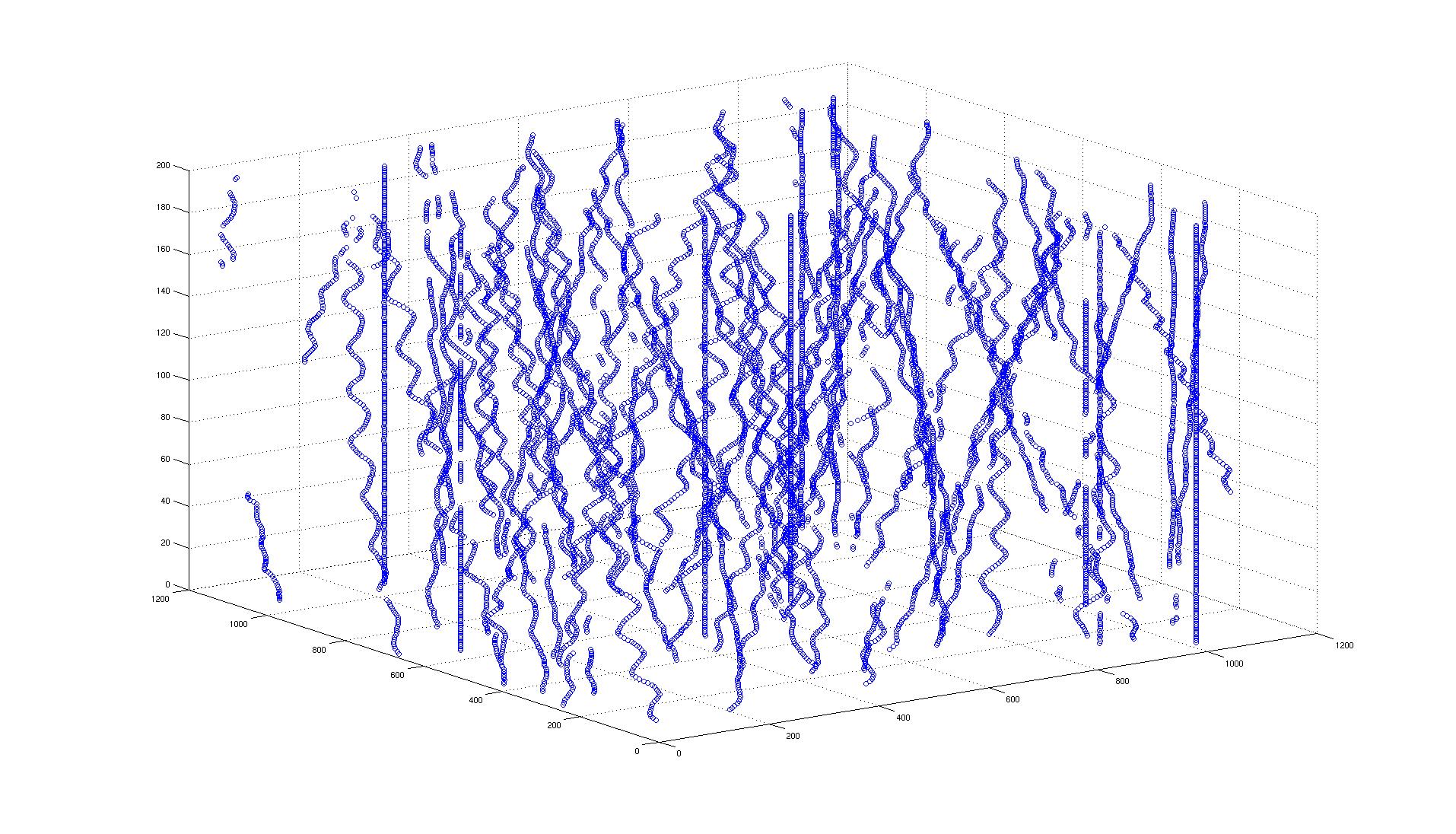
Spermatozoid tracking
Participants : Clarens Caraccio, Xavier Descombes.
In this work, we have proposed an algorithm for tracking spermatozoid in a sequence of confocal images. We first detect the spermatozoids by thresholding the result of a top hat operator. The thresold is automatically estimated using Otsu's method. We then analyse the different connected components to detect overlaps between adjacent spermatozoids. Temporal neighbors are selected based on the spatial consistency of the object sets between two consecutive time. A first result is given on figure 9 .